
En France, la maladie touche environ 1 naissance sur 60 000 chaque année et un peu plus de 500 malades sont recensés. La maladie de Gaucher (de type 1) survient plus souvent chez les personnes d’origine juive ashkénaze (jusqu’à 1/1 000 nouveaux cas par an), tandis que de nombreux cas de type 3 ont été́ décrits dans la région de Norrbotten, en Suède.
La maladie de Gaucher est une maladie génétique ; les patients atteints ont une mutation du gène aboutissant à un déficit de l’activité d’une enzyme (2) lysosomale : la glucocérébrosidase dont la conséquence est l’accumulation du glucocérébroside.
Les manifestations de la maladie de Gaucher sont très variables, ce qui a conduit à classer la maladie en trois types selon l’âge d’apparition et la gravité :
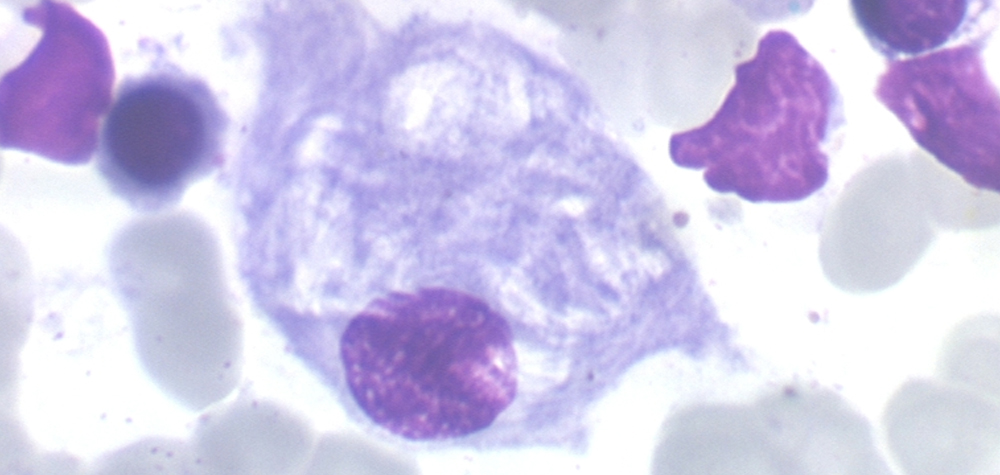
L’accumulation de glucocérébroside dans les lysosomes (3) affecte principalement les macrophages (4), chargés de phagocyter (ou « manger ») les substances étrangères à l’organisme. Ces macrophages pleins de glucocérébroside, deviennent très volumineux (appelés «cellules de Gaucher») et sont responsable de l’augmentation du volume de certains organes (foie, rate, os et parfois poumons).
Dans le foie, il en résulte une mort cellulaire (nécrose) qui est remplacé par une cicatrice rigide et fibreuse pouvant provoquer une cirrhose et un mauvais fonctionnement du foie. L’infarctus osseux entraine une nécrose de l’os (ostéonécrose) qui peut se localiser sur la tête du fémur et conduire progressivement vers la destruction de l’articulation (arthrose). Si la fibrose (5) concerne le poumon, elle conduit à une difficulté à respirer (dyspnée).
Les personnes atteintes de la maladie de Gaucher de type 1 sont souvent fatiguées et ont fréquemment le ventre gonflé, du fait de l’augmentation de volume du foie et de la rate. Elles saignent souvent du nez et des gencives. Des bleus (hématomes, ecchymoses) apparaissent facilement, parfois au moindre contact et sans traumatisme important. Les saignements plus importants sont rares. Ces anomalies sanguines sont en général liées au dysfonctionnement du foie et le la rate.
Les personnes peuvent avoir des douleurs osseuses invalidantes. Cependant, tous les malades n’ont pas forcément toutes les manifestations décrites, et certaines n’auront aucun symptôme. Les manifestations peuvent diminuer ou disparaitre si un traitement est donné assez tôt.
Les personnes atteintes de la maladie de Gaucher de type 1 mènent le plus souvent une vie très proche de la « normale ». Les enfants peuvent aller à l’école et les adultes avoir une activité professionnelle. Cela étant, plusieurs manifestations de la maladie ont des conséquences sur la vie quotidienne, et parmi elles, la fatigue.
Le dosage enzymatique est l’outil qui permet le diagnostic de la maladie de Gaucher dans la plupart des cas. Ce test, réalisé à partir d’une prise de sang, mesure l’activité de la glucocérébrosidase. Chez les personnes malades, l’activité de cette enzyme est très faible.
Ce test ne permet pas de distinguer les 3 formes de la maladie.
Le test génétique, à partir d’une prise de sang, n’est pas nécessaire pour poser le diagnostic de la maladie de Gaucher mais il peut permettre en cas de doute de déterminer le type de maladie. La réalisation de ce test peut conduire à la réalisation d’études génétiques chez d’autres membres de la famille.
Une fois le diagnostic posé, la recherche des signes habituellement observés dans la maladie de Gaucher est entreprise pour déterminer le degré de gravité de la maladie. Ces examens vont permettre une prise en charge adaptée à chaque malade (bilan sanguin, examens d’imagerie de l’abdomen et des os…)
La question du risque de transmission de la maladie est inévitablement évoquée. Il est généralement proposé au conjoint de faire un examen sanguin s’il y a consanguinité́ ou si le conjoint est d’origine juive ashkénaze et, dans les autres cas, selon le désir du couple. Le test détermine si le conjoint est lui aussi porteur de la ou des anomalie(s) génétique(s).
Par ailleurs, il est recommandé́ de discuter avec son médecin de tout désir d’enfant car le traitement éventuel aura besoin d’être adapté.
Le traitement enzymatique substitutif (l’imiglucérase) est utilisé́ pour traiter les malades atteins de la maladie de Gaucher de type 1. Ce traitement consiste à administrer directement une enzyme identique à la glucocérébrosidase dans le sang afin de rétablir un niveau d’activité́ enzymatique suffisant et éliminer en partie la substance accumulée.
Ce traitement doit être poursuivi à vie. Il est donné par voie intraveineuse une fois tous les 15 jours. Ce traitement a considérablement amélioré l’état des personnes atteintes.
Ce traitement est également proposé à des enfants et adolescents atteints de la maladie de Gaucher de type 3 qui ne présentent pas de manifestations neurologiques graves irréversibles.
Lorsque l’imiglucérase ne peut pas être administrée, ou en cas de contre-indication de l’imiglucérase, un autre médicament peut servir d’alternative. Ce traitement qui se prend par la bouche à pour objectif de réduire la quantité́ de la substance accumulée dans les cellules. Néanmoins, ce traitement est moins efficace que le traitement substitutif.
D’autres traitements sont nécessaires visant à prendre en charge les différentes manifestations de la maladie (transfusion, traitement des complications osseuses…). Une greffe de moelle osseuse peut être indiquée pour un très petit nombre de personnes présentant une forme très sévère de la maladie (type 3).
La fatigue diminue en 3 mois et disparait généralement à 6 mois. Les anomalies sanguines s’atténuent en 6 à 12 mois. Les crises douloureuses des os diminuent chez 9 personnes sur 10 mais l’amélioration des manifestations au niveau des os est généralement un peu plus lente (jusqu’à deux ans). Certaines anomalies visibles à la radiographie du squelette et sur les IRM ne disparaissent pas sous traitement. Une diminution des douleurs et de la taille du foie est observée chez 3 à 4 personnes sur 10 et la diminution du volume de la rate est observée chez 5 à 6 personnes sur 10 traitées.
(1) Glucocérébroside : cérébroside dont le sucre constitutif est une molécule de glucose liée par une liaison β-osidique à un céramide. Ce glycolipide, présent dans les membranes cellulaires animales, est aussi la première étape de la biosynthèse des autres sphingosidolipides et également la dernière de leur dégradation, avant l’hydrolyse de la liaison osidique par une bêta-glucosidase spécifique (glucocérébrosidase ou glucosylcéramide-bêta-glucosidase). L’accumulation dans la rate et le foie, qui caractérise la maladie de Gaucher, est due à un défaut de cette bêta-glucosidase.
(2) Enzymes : substance de nature protéinique, généralement macromoléculaire, douée d’une activité catalytique vis-à-vis de molécules, appelées substrats. La plupart des réactions biochimiques sont catalysées par des enzymes spécifiques.
(3) Lysosomes : organite intracellulaire d’environ 0,5 µm limité par une membrane, qui renferme des enzymes hydrolytiques. De nombreuses maladies congénitales sont dues à un défaut d’enzymes lysosomiques. Les lysosomes sont responsables de la destruction cellulaire et de la digestion des bactéries.
(4) Macrophages : cellule monocytaire dont le précurseur est issu de la moelle osseuse et du sang circulant, riche en enzymes lysosomiaux et possédant des capacités de phagocytose. Il est présent dans tout tissu (appelé cellule de Kuppfer dans le foie). Il joue un rôle important dans l’immunité.
(5) Fibrose du foie : (Vient du latin : fibra : fibre ; ose : processus chronique) Lésion non spécifique, caractérisée par une hyperplasie du tissu conjonctif, par multiplication des fibroblastes et augmentation de la synthèse des fibres collagènes et/ou élastiques